Monarch KG Build Process
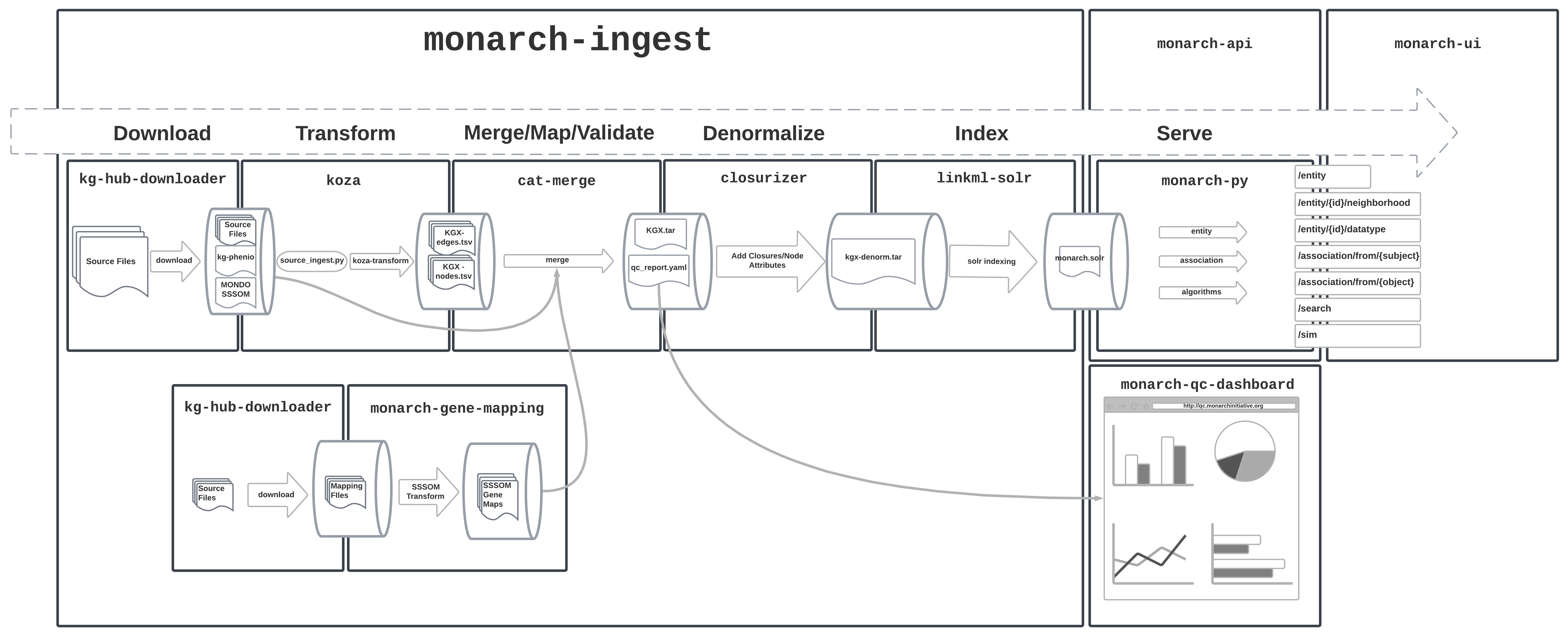
Download
A weekly job indepent from the KG build process runs to download data sources and store then on a cloud bucket. This replaces DipperCache from the old pipeline. KGHub Downloader reads from downloads.yaml to download each file. Some post-processing is done in a shell script before the files are uploaded to the cloud bucket.
At the start of the main ingest build, data files are copied from the cloud bucket.
Transform
A call to the ingest command line tool runs each source ingest defined in ingest.yaml, producing both KGX tsv and RDF nt output.
Source Ingests
Ingests are documented individually in the Sources section of this documentation. Ingests are either node or edge specific, and use IDs as defined in the source data files without additional re-mapping of identifiers. The primary role they have is to represent sources in biolink model and KGX format, and secondarily they may also subset from the source files. The output of individual ingests can be found in the transform_output directory in each release.
Phenio-KG
Ontologies in Monarch are built first as Phenio, then converted into the biolink model and represented as KGX in kg-phenio.
The ingest CLI has transform_phenio method then performs some further filtering on the kg-phenio node and edge files. Limiting to nodes and edges that match a subset of curie namespaces, and limiting node property columns to a relevant subset.
Merge
With all transforms complete, the individual kgx node and edge files in output/transform_output can be combined into a merged graph. This is done by the merge command in the ingest CLI.
At this point, the individual node and edge KGX files from the transforms may not have matching IDs, and in fact, we may have edges that point to nodes that are not present in our canonical node sources (e.g. a STRING edge that points to an ENSEMBL gene that can't be mapped to HGNC).
The merge process is broken down into join, normalize, and prune steps, implemented using koza graph operations.
Join
The first step loads all node KGX files into a single nodes table and all edge KGX files into a single edges table in DuckDB.
Normalize
The normalize step replaces subject and object IDs in edge files using SSSOM mapping files, with the IDs from the initial ingests stored in original_subject and original_object fields.
Mappings for genes are generated in our monarch-gene-mapping process, and are available at data.monarchinitiative.org.
Diseases are mapped using the MONDO SSSOM.
This step requires that the subject of the SSSOM file be our canonical ID, and the object be the non-canonical ID. There is room for improvement here.
Prune
After edges have been normalized, it's important to cull edges that point to nodes that don't exist in the graph. The prune step performs joins against the node table to split out these edges into their own table (dangling_edges) that can be used for QC purposes.
A group of edges that wind up in this file could be due to a number of reasons: * We're missing an ontology or other node source that is required for an ingest/source: this is something we want to fix 👎 * We're missing mapping necessary to translate between an edge ingest and our canonical node sources: this is something we want to fix 👎 * The edge ingest includes edges which can't be mapped to our canonical node sources: this is a feature! 👍
We have a visualization of this split between connected and dangling edges for each ingest on our QC Dashboard that we can use to problem-solve our mappings and node sources.
Neo4j
A neo4j dump is created using the merged tar.gz file using KGX's neo4j loader and a docker container. This process is defined directly in the Jenkinsfile.
Denormalize
For Solr (and secondarily SQLite) we produce a denormalized edge file, which includes additional details for the subjects and objects of each edge, including the category, namespace/prefix, and ontology ancestor closures following the GOLR pattern (ID and label closure lists). The closure file is generated by relation-graph and are included in the kg-phenio download. The after_download script makes a filtered version that only includes rdfs:subClassOf, BFO:0000050, and UPHENO:0000001.
SQLite
A SQLite database file is produced by loading node and edge files into a SQLite database using a simple shell script, along with the primary node and edge tables, edge tables for danging and denormalized edges are included as well.
Solr
Our solr index is loaded directly from the node kgx tsv file and the denormalized edge tsv file using LinkML-Solr. The LinkML schema for the Solr index is lives in the monarch-py data access library (see documentation for Entity and Association classes).
LinkML-Solr starts Solr in docker via the lsolr command, defines the Solr schema based on the LinkML Schema and then bulk loads the data. Currently, a small amount of additional Solr configuration (defining new field types, and copy-fields declarations to fill them) is done via curl commands in shell scripts.
Our solr load process is defined in scripts/load_solr.sh