Input data
Each GPSEA analysis starts with the ingest of genotype and phenotype data. There are many things to be done. We need to check the HPO terms to point out any logical inconsistencies or to remove redundancies, annotate the variants with respect to genes and transcripts, or check uniqueness of the identifiers of the cohort members. Additionally, most GPSEA analyses need an information about the “anatomy” of the transcript and/or protein corresponding to the gene(s) of interest.
This section will explain how to ingest the phenopackets, how to persist the cohort for later, to save the preprocessing time, and how to get the transcript and protein information.
Create a cohort
GPSEA workflow starts with a collection
of GA4GH Phenopacket Schema phenopackets
which are transformed into a Cohort.
Load HPO
The Q/C requires access to Human Phenotype Ontology, which we can load using the hpo-toolkit:
>>> import hpotk
>>> store = hpotk.configure_ontology_store()
>>> hpo = store.load_minimal_hpo(release='v2024-07-01')
The code downloads the HPO data from GitHub and stores it in the user’s home directory,
to speed up the analysis by downloading the ontology data only once.
The latest HPO can be loaded by leaving out the release argument.
Note
If storing HPO locally is not desired, HPO can also be loaded directly from GitHub, at the expense of additional network traffic:
release = 'v2024-07-01'
hpo = hpotk.load_minimal_ontology(f'https://github.com/obophenotype/human-phenotype-ontology/releases/download/{release}/hp.json')
Cohort creator
The CohortCreator is responsible for transforming
phenopackets into a Cohort.
The transformation is very flexible, with an array of parameters to tweak.
However, the majority of cases should be covered by the default settings
and we recommend to configure the default cohort creator with
the configure_caching_cohort_creator() method:
>>> from gpsea.preprocessing import configure_caching_cohort_creator
>>> cohort_creator = configure_caching_cohort_creator(hpo)
The cohort_creator will check the HPO terms for any invalid or obsolete
terms, as well as point out any logical inconsistencies.
The functional annotation and validation of variants is delegated to
Variant Effect Predictor and Variant Validator REST APIs,
and the API responses are cached in the current working directory, to reduce the network bandwith.
The cohort is also checked for individuals with non-unique ID.
Load phenopackets
We can create a cohort starting from a collection of Phenopacket objects
provided by Python Phenopackets library.
For the purpose of this example, we will load a cohort of patients with pathogenic mutations in RERE gene
which are included in the release 0.1.18 of Phenopacket Store.
We use Phenopacket Store Toolkit
(ppktstore in the code) to reduce the boilerplate code
associated with extracting phenopacket data from Phenopacket Store release:
>>> from ppktstore.registry import configure_phenopacket_registry
>>> registry = configure_phenopacket_registry()
>>> with registry.open_phenopacket_store(release="0.1.19") as ps:
... phenopackets = tuple(ps.iter_cohort_phenopackets('RERE'))
>>> len(phenopackets)
19
The code creates a registry which will download the ZIP file
for a given Phenopacket Store data release and store it in user’s home directory.
Then, phenopackets of the RERE cohort are extracted,
resulting in a tuple with 19 phenopackets.
The actual transformation of the phenopackets into a Cohort
is orchestrated by the load_phenopackets()
loader function:
>>> from gpsea.preprocessing import load_phenopackets
>>> cohort, qc_results = load_phenopackets(
... phenopackets=phenopackets,
... cohort_creator=cohort_creator,
... )
Individuals Processed: ...
>>> len(cohort)
19
The loader applies the CohortCreator to phenopackets, while keeping track of any issues,
and we get back a cohort as well as a PreprocessingValidationResult (qc_results)
with any Q/C issues.
The summarize() method summarizes the found issues:
>>> qc_results.summarize()
Validated under permissive policy
No errors or warnings were found
No issues were found in the current cohort.
Note
See Quality control (few paragraphs below) for more info on qc_results.
Alternative phenopacket sources
More often than not, the phenopackets of interest will not be deposited in Phenopacket Store. For these use cases, GPSEA simplifies loading phenopackets from a list of JSON files, or from a folder with phenopacket JSON files.
The load_phenopacket_files() function loads
phenopackets from one or more paths that point to phenopacket JSON files:
>>> from gpsea.preprocessing import load_phenopacket_files
>>> pp_file_paths = ('path/to/phenopacket1.json', 'path/to/phenopacket2.json')
>>> cohort, qc_results = load_phenopacket_files(pp_file_paths, cohort_creator)
Alternatively, you can load an entire directory of phenopackets
with the load_phenopacket_folder() loader function.
Starting with path to a directory with phenopacket JSON files,
the loader includes all files that end with *.json suffix
and ignores any other files or sub-directories:
>>> from gpsea.preprocessing import load_phenopacket_folder
>>> pp_dir = 'path/to/folder/with/many/phenopacket/json/files'
>>> cohort, qc_results = load_phenopacket_folder(pp_dir, cohort_creator)
Quality control
Besides the Cohort, the loader functions also provide Q/C results (qc_results)
as PreprocessingValidationResult.
The Q/C checker points out as many issues as possible (not just the first one),
to address all issues at once, as opposed to time-consuming iterative fixing.
The issues can be explored programmatically
through the PreprocessingValidationResult API,
or we can display a summary with the
summarize() method:
>>> qc_results.summarize()
Validated under permissive policy
No errors or warnings were found
In this case, no Q/C issues were found.
Persist the cohort for later
The preprocessing of a cohort can take some time even if we cache the responses from remote resources,
such as Variant Effect Predictor, Variant Validator, or Uniprot.
GPSEA ships with a custom encoder and decoder
that integrates with Python’s built-in json module,
to persist a Cohort to a JSON file on Friday afternoon,
and load it back on Monday morning.
Example
We can dump the Cohort into JSON
by providing GpseaJSONEncoder via the cls option to the json module functions,
such as the json.dumps() which encodes an object into a JSON str:
>>> import json
>>> from gpsea.io import GpseaJSONEncoder
>>> encoded = json.dumps(cohort, cls=GpseaJSONEncoder)
>>> encoded[:80]
'{"members": [{"labels": {"label": "Subject 8", "meta_label": "PMID_29330883_Subj'
Here we see the first 80 letters of the JSON object.
We can decode the JSON object with GpseaJSONDecoder to get the same cohort back:
>>> from gpsea.io import GpseaJSONDecoder
>>> decoded = json.loads(encoded, cls=GpseaJSONDecoder)
>>> cohort == decoded
True
We will leave persisting the cohort into an actual file or another data store as an exercise for the interested readers.
Choose the transcript and protein of interest
G/P association analysis is performed with reference to a specific gene transcript and the corresponding protein sequence. The transcript and protein information is also needed for summary statistics generation, to visualize variant distribution with respect to transcript/protein sequence, and in most genotype partitioning schemes.
Here we point out our recommendations for choosing the transcript of interest and let GPSEA fetch the associated data.
Choose the transcript
For the analysis, the MANE transcript (i.e., the “main” biomedically relevant transcript of a gene) should be chosen unless there is a specific reason not to (which should occur rarely if at all).
A good way to find the MANE transcript is to search on the gene symbol (e.g., TBX5) in ClinVar and to choose a variant that is specifically located in the gene. The MANE transcript will be displayed here (e.g., NM_181486.4(TBX5):c.1221C>G (p.Tyr407Ter)). The RefSeq identifier of the encoded protein (e.g. NP_852259.1 for TBX5) should be also readily available on the ClinVar website:
>>> tx_id = "NM_181486.4"
>>> pt_id = "NP_852259.1"
Get the transcript data
Besides the transcript accession ID, the downstream analysis may need more information
about the “anatomy” of the transcript of interest, such as the coordinates of the exons
or the untranslated regions.
The coordinates can be represented either in GRCh37
or GRCh38 (recommended) reference genomes.
GPSEA models the transcript anatomy with
TranscriptCoordinates class,
and there are several ways to prepare the transcript coordinates,
which we list here by their convenience in decreasing order.
Fetch transcript coordinates from Variant Validator REST API
The most convenient way for getting the transcript coordinates is to use
the REST API of the amazing Variant Validator.
GPSEA simplifies querying the API and parsing the response
with a TranscriptCoordinateService.
We use configure_default_tx_coordinate_service() to get a service
that caches the response locally to prevent repeated API queries for the same transcript accession:
>>> from gpsea.preprocessing import configure_default_tx_coordinate_service
>>> txc_service = configure_default_tx_coordinate_service(genome_build="GRCh38.p13")
Now we can fetch the coordinates of the MANE transcript of TBX5 on GRCh38:
>>> tx_coordinates = txc_service.fetch(tx_id)
>>> tx_coordinates.identifier
'NM_181486.4'
Provide the transcript coordinates manually
TODO: implement and document!
Showcase transcript data
Based on tx_coordinates, GPSEA knows about the location of the transcript region in the reference genome:
>>> tx_coordinates.region.contig.name
'12'
>>> tx_coordinates.region.start
18869165
>>> tx_coordinates.region.end
18921399
>>> tx_coordinates.region.strand.symbol
'-'
or the count and coordinates of exons:
>>> len(tx_coordinates.exons)
9
>>> print(tx_coordinates.exons[0]) # coordinate of the 1st exon
GenomicRegion(contig=12, start=18869165, end=18869682, strand=-)
Note
The regions spanned by transcripts, exons, UTRs, as well as by variants
are represented either as a GenomicRegion
or a Region.
Furthermore, we know that the transcript is coding
>>> tx_coordinates.is_coding()
True
and so we can see that 8 exons include protein coding sequences,
>>> len(tx_coordinates.get_cds_regions())
8
2 exons include 5’ untranslated regions,
>>> len(tx_coordinates.get_five_prime_utrs())
2
and the coding sequence includes 1554 coding bases and 518 codons.
>>> tx_coordinates.get_coding_base_count()
1554
>>> tx_coordinates.get_codon_count()
518
Fetch protein data
Specific domains of a protein may be associated with genotype-phenotype correlations. For instance, variants in the pore domain of PIEZO1 are associated with more severe clinical manifestions in dehydrated hereditary stomatocytosis Andolfo et al., 2018.
GPSEA uses the protein data in several places: to show distribution of variants with respect to the protein domains
or other features of biological interest, and to group the individuals based on presence of a variant predicted
to affect the protein features.
In all cases, the protein data must be formatted as an instance of ProteinMetadata
and here we show how to get the data and use it in the analysis.
The protein data (ProteinMetadata) can be obtained in several ways,
ordered (again) by their convenience:
fetched from UniProt REST API
parsed from a JSON file downloaded from UniProt
entered manually from a data frame
Fetch data from UniProt REST API
The most convenient way to obtain the protein data is to use a ProteinMetadataService.
We recommend using the configure_default_protein_metadata_service()
to reduce the amount of the associated boiler-plate code:
>>> from gpsea.preprocessing import configure_default_protein_metadata_service
>>> pms = configure_default_protein_metadata_service()
Then, fetching the data for protein accession NP_852259.1 encoded by the NM_181486.4 transcript of TBX5 is as simple as running:
>>> protein_meta = pms.annotate(pt_id)
>>> protein_meta.protein_id
'NP_852259.1'
>>> protein_meta.protein_length
518
>>> len(protein_meta.protein_features)
5
The protein_meta represents the TBX5 isoform that includes 518 aminoacids and two features of interest, which we can see on the following screenshot of the UniProt entry for TBX5:
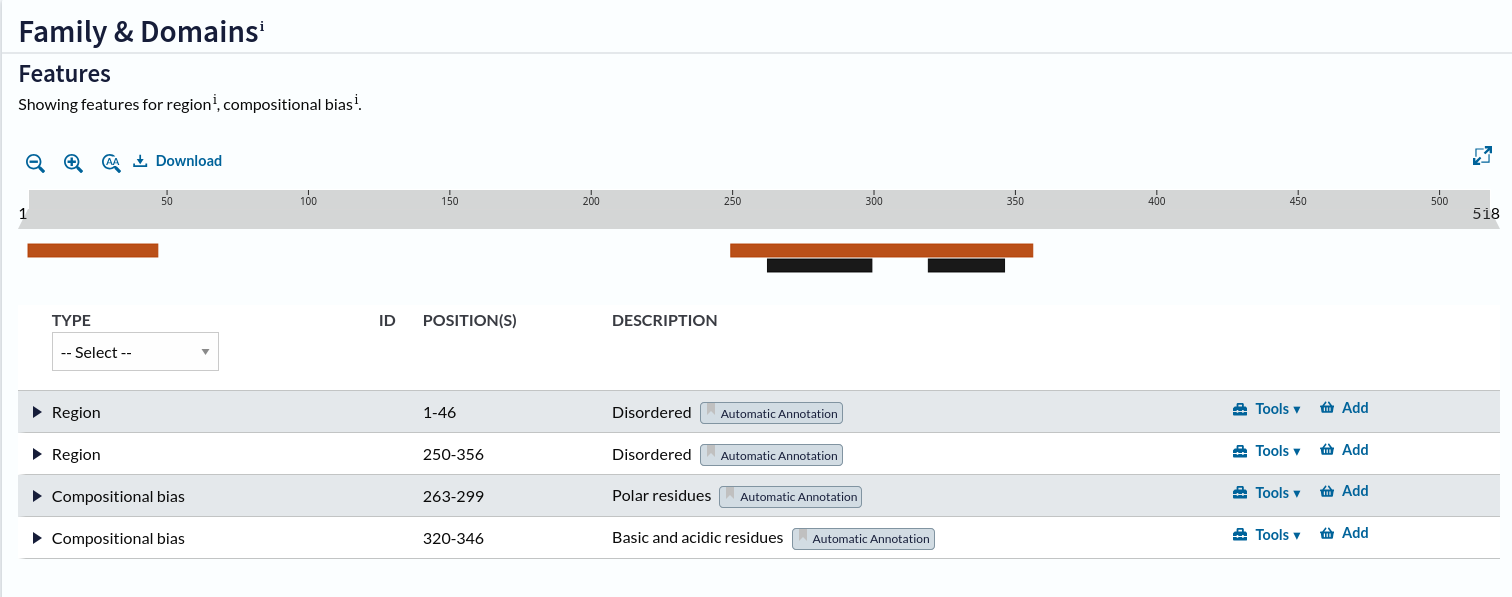
Protein features of TBX5 (Q99593, UniProt entry)
UniProt shows four protein features:
the Disordered region (1-46)
the Disordered region (250-356)
presence of Polar residues (263-299)
presence of Basic and acidic residues (320-346).
Parse UniProt JSON dump
In the cases, when the REST API cannot give us the data for a protein of interest,
we can download a JSON file representing the protein features manually,
and load the file into ProteinMetadata.
To do this, click on the Download symbol (see the UniProt screenshot figure above). This will open a dialog
that allows the user to choose the contents of the JSON file.
Do not change the default option (Features - Domain, Region).
Provided that the file has been saved as docs/user-guide/data/Q99593.json,
the ProteinMetadata can be loaded using from_uniprot_json() function.
Note that you will need to obtain information about the protein name (label)
and protein_length, but these are shown in the UniProt entry:
>>> from gpsea.model import ProteinMetadata
>>> downloaded_uniprot_json = "docs/user-guide/data/Q99593.json"
>>> protein_meta = ProteinMetadata.from_uniprot_json(
... protein_id="NP_852259.1",
... label="transforming growth factor beta receptor 2",
... uniprot_json=downloaded_uniprot_json,
... protein_length=518,
... )
Enter features manually
The information about protein features provided by UniProt entries may not always be complete. Here we show how to enter the same information manually, in a custom protein dataframe.
The frame can be created e.g. by running:
>>> import pandas as pd
>>> domains = [
... {"region": "Disordered","category": "region", "start": 1, "end": 46, },
... {"region": "Disordered", "category": "region", "start": 250, "end": 356, },
... {"region": "Polar residues", "category": "compositional bias", "start": 263, "end": 299, },
... {"region": "Basic and acidic residues", "category": "compositional bias", "start": 320, "end": 346, },
... ]
>>> df = pd.DataFrame(domains)
The ProteinMetadata is then created using from_feature_frame() function:
>>> protein_meta = ProteinMetadata.from_feature_frame(
... protein_id="NP_852259.1",
... label="transforming growth factor beta receptor 2",
... features=df,
... protein_length=518,
... )