Group by allele count
Sometimes, we may want to compare individuals with different allele counts of a single variant category.
For instance, we may want to compare the survival of individuals harboring a mutation in EGFR (\(AC = 1\)) with those with no such mutation (\(AC = 0\)). Alternatively, in some genes, heterozygous mutations (\(AC = 1\)) and biallelic mutations (\(AC = 2\)) can lead to different diseases.
It is worth pointing out the difference of the allele count analysis with the variant category analysis described in the Group by variant category section. The allele count analysis compares the allele counts while keeping the variant type constant, whereas the variant category analysis compares variant types while keeping the total allele count constant.
A classifier for the allele count analysis is created with
the allele_count() function.
The function needs two arguments:
target with a VariantPredicate for selecting the target variant type,
and counts with the target allele counts.
The target variant predicate typically selects a broad variant types - for instance, all variants that affect a gene of interest.
The counts take the allele counts to compare.
Let’s show a few examples.
Examples
Compare the individuals with EGFR mutation
We can use the allele count analysis to test for G/P associations between the individuals with no mutation in EGFR and the individuals harboring 1 variant allele.
First, let’s create a VariantPredicate
to select variants that affect EGFR:
>>> from gpsea.analysis.predicate import gene
>>> affects_egfr = gene(symbol="EGFR")
>>> affects_egfr.description
'affects EGFR'
Next, we create allele count classifier to partition the individuals based on presence of zero or one EGFR mutation allele:
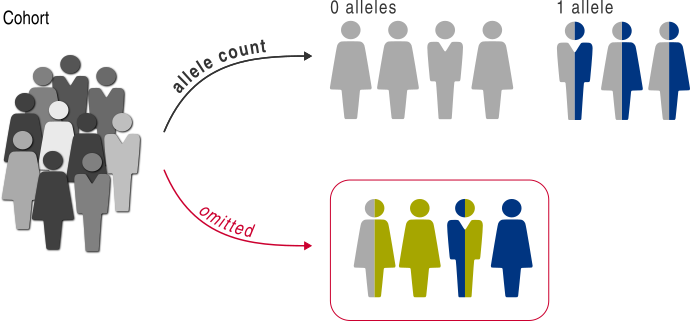
>>> from gpsea.analysis.clf import allele_count
>>> gt_clf = allele_count(
... counts=(0, 1),
... target=affects_egfr,
... )
>>> gt_clf.class_labels
('0 alleles', '1 allele')
The allele_count needs two inputs.
The counts takes a tuple of the target allele counts,
in order to partition the individuals based on zero or one allele.
The target takes a VariantPredicate
to select the target variants.
The target is particularly important
if the cohort members include variants in other genes than EGFR.
The resulting gt_clf can partition a cohort along the genotype axis,
e.g. to compare the patient survivals in a survival analysis <survival>.
Compare the individuals with monoallelic and biallelic mutations
As another example, let’s partition individuals based on one or two alleles of a target mutation.
For this example, the target mutation is any mutation that affects LMNA:
>>> from gpsea.analysis.predicate import gene
>>> affects_lmna = gene(symbol="LMNA")
>>> affects_lmna.description
'affects LMNA'
and we will compare the individuals with one allele with those with two alleles:
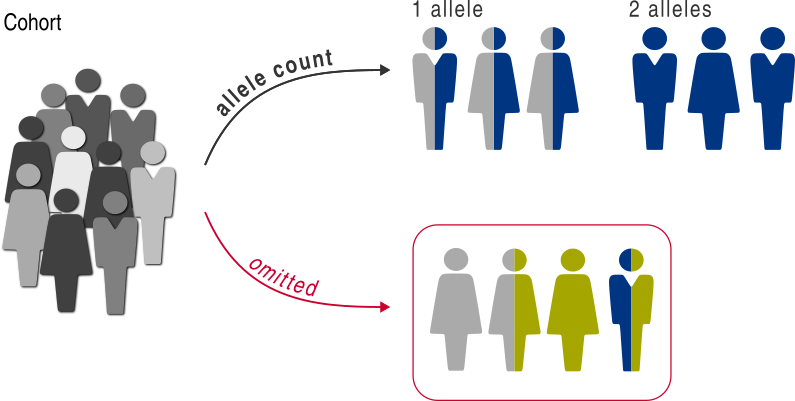
>>> gt_clf = allele_count(
... counts=(1, 2),
... target=affects_lmna,
... )
>>> gt_clf.class_labels
('1 allele', '2 alleles')
The classifier assigns the individuals into one of two classes: those with one LMNA variant allele and those with two LMNA variant alleles. Any cohort member with other allele counts (e.g. 0 allele or 3 alleles) is ignored.